INTRODUCING NITRILES
This page explains what nitriles are and looks at their simple physical
properties such as solubility and boiling points.
What are nitriles?
Nitriles contain the -CN group, and used to be known as cyanides.
Some simple nitriles
The smallest organic nitrile is ethanenitrile, CH3CN, (old name:
methyl cyanide or acetonitrile - and sometimes now called ethanonitrile).
Hydrogen cyanide, HCN, doesn't usually count as organic, even though it
contains a carbon atom.
Notice the triple bond between the carbon and nitrogen in the -CN group.
The three simplest nitriles are:
CH3CN
|
ethanenitrile
|
CH3CH2CN
|
propanenitrile
|
CH3CH2CH2CN
|
butanenitrile
|
When you are counting the length of the carbon chain, don't forget the
carbon in the -CN group. If the chain is branched, this carbon usually counts
as the number 1 carbon
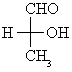
Note: Compounds like this are formed when aldehydes react with hydrogen cyanide.
This is therefore the sort of branched nitrile that you are most likely to come
across at this level.
Physical properties
Boiling points
The small nitriles are liquids
at room temperature.
nitrile
|
boiling point (°C)
|
CH3CN
|
82
|
CH3CH2CN
|
97
|
CH3CH2CH2CN
|
116 - 118
|
Note: The majority of the data sheets I have looked at quote this boiling range
for butanenitrile. I don't know why it doesn't seem to have a precise boiling
point.
These boiling points are very high for the size of the molecules - similar
to what you would expect if they were capable of forming hydrogen bonds.
However, they don't form hydrogen bonds - they don't have a hydrogen
atom directly attached to an electronegative element.
They are just very polar molecules. The nitrogen is very electronegative
and the electrons in the triple bond are very easily pulled towards the
nitrogen end of the bond.
Nitriles therefore have strong permanent dipole-dipole attractions as well
as van der Waals dispersion forces between their molecules.
Solubility in water
Ethanenitrile is completely soluble in water, and the solubility then falls
as chain length increases.
nitrile
|
solubility at 20°C
|
CH3CN
|
miscible
|
CH3CH2CN
|
10 g per 100 cm3 of water
|
CH3CH2CH2CN
|
3 g per 100 cm3 of water
|
The reason for the solubility is that although nitriles can't hydrogen bond
with themselves, they can hydrogen bond with water molecules.
One of the slightly positive hydrogen atoms in a water molecule is
attracted to the lone pair on the nitrogen atom in a nitrile and a hydrogen
bond is formed.
There will also, of course, be dispersion forces and dipole-dipole
attractions between the nitrile and water molecules.
Forming these attractions releases energy. This helps to supply the energy
needed to separate water molecule from water molecule and nitrile molecule from
nitrile molecule before they can mix together.
As chain lengths increase, the hydrocarbon parts of the nitrile molecules
start to get in the way.
By forcing themselves between water molecules, they break the relatively
strong hydrogen bonds between water molecules without replacing them by
anything as good. This makes the process energetically less profitable, and so
solubility decreases.
Hydrolysis
of nitriles with aqueous acid to give carboxylic acids
Description: Addition of water and acid to a
nitrile leads to formation of a carboxylic acid.
Notes:
- This reaction is referred to as
“acidic hydrolysis”.
- The reaction is generally used
with water as solvent, so an excess of water is present. The acid used is
often written as “H3O(+)”
Protonation of the nitrile nitrogen by acid (Step 1, arrows A and B)
makes the nitrile carbon a better electrophile. Attack at the carbon by
water (Step 2, arrows C and D) followed by proton transfer (Step 3,
arrows E and F) gives a species that is inesonance with a protonated
amide (arrows G and H). Addition of water to the protonated amide (Step
4, arrows I and J) followed by proton transfer (Step 5, arrows K and L)
result in formation of NH3(+) which is an excellent leaving group.
Expulsion of NH3 through 1,2-addition (Step 6, arrows M and N) followed
by deprotonation (Step 7, arrows O and P) give the carboxylic acid.
Reaction type: Nucleophilic Acyl Substitution then
Nucleophilic Addition
Summary:
- Nitriles,
RCºN, react with Grignard reagents or
organolithium reagents to give ketones.
- The
strongly nucleophilic organometallic reagents add to the CºN bond in a similar fashion to
that seen for aldehydes
and ketones.
- The
reaction proceeds via an imine salt intermediate that is then hydrolyzed to give
the ketone product.

- Since
the ketone is not formed until after the addition of water,
the organometallic reagent does not get the opportunity to react with
the ketone product.
Dehydration
of amides to give nitriles
Description: Primary amides can be
converted to nitriles with a dehydrating reagent such as P2O5
.
Notes: Note that the net effect of this reaction is to
remove two H atoms and one O from the amide. For this reason this is called a “dehydration”.
Only primary amides work for this reaction.
Other reagents can be used for this, however, such as thionyl chloride (SOCl2)
Examples:
Notes:
Mechanism:
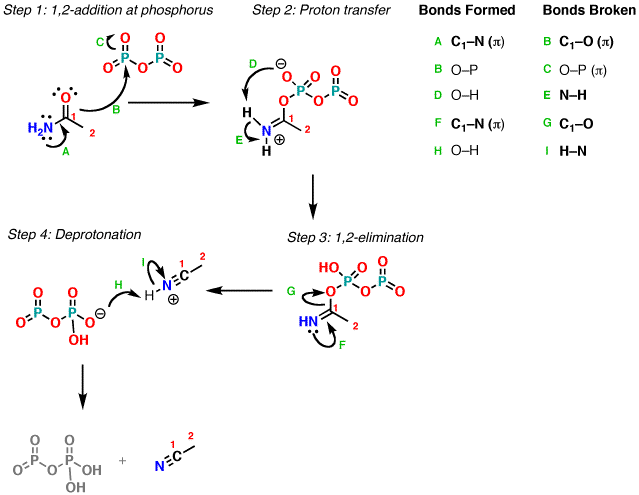
The reaction begins with the oxygen of the amide
attacking phosphorus (through a resonance form) forming an O–P bond (Step 1,
arrows A, B, and C). After a proton transfer (Step 2, arrows D and E) a lone
pair from nitrogen forms a new C–N bond, expelling oxygen (Step 3, arrows F and
G). Finally the nitrogen is deprotonated (Step 4, arrows H and I) to give the
neutral nitrile.
Notes:
There are certainly other reasonable ways to
draw proton transfer (Step 2) as well as other bases to use for deprotonation
(Step 4) besides phosphate. This is just one reasonable possibility.
It’s also reasonable to show fragmentation of
the P–O–P bond in step 3, although for simplicity’s sake this was not drawn.
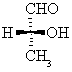








{kind=link}
{kind=link}